Software
ASCAT is a method to derive copy number profiles of tumor cells, accounting for normal cell admixture and tumor aneuploidy. ASCAT infers tumor purity (the fraction of tumor cells) and ploidy (the amount of DNA per tumor cell, expressed as multiples of haploid genomes) from SNP array or massively parallel sequencing data, and calculates whole-genome allele-specific copy number profiles (the number of copies of both parental alleles for all SNP loci across the genome).
The latest ASCAT version is available as an R package on GitHub at https://github.com/VanLoo-lab/ascat. Instructions to install and try the software are provided on the GitHub page.
Running ASCAT
In its simplest form (with matched normal data available, without GC wave correction and all samples female), ASCAT can be run as follows:
library(ASCAT)
ascat.bc = ascat.loadData("Tumor_LogR.txt", "Tumor_BAF.txt", "Germline_LogR.txt", "Germline_BAF.txt")
ascat.plotRawData(ascat.bc)
ascat.bc = ascat.aspcf(ascat.bc)
ascat.plotSegmentedData(ascat.bc)
ascat.output = ascat.runAscat(ascat.bc)
The ascat.loadData function by default assumes all samples are female. An extra optional parameter (gender = …) allows setting the gender of samples (in vector format, using "XX" for females and "XY" for males).
ASCAT can be run in different modes: without matched normal data, with a logR correction (GC content and replication timing), with a multi-segmentation and on high-throughput sequencing (HTS) data. Examples to run ASCAT using such different modes can be found here.
Input data formats and supported platforms
1) SNP arrays
ASCAT is platform and species-independent and works for both Illumina and Affymetrix SNP arrays. The input required includes matrices of LogR and B Allele Frequency (BAF) data (rows are probes or SNP loci and columns are samples). ASCAT requires identically formatted LogR and BAF files for both tumor and germline data (with matching samples on matching rows in all four files). For examples of the precise data format, see our simulated example data (7.62 MB, zip).
Input data for ASCAT can be obtained directly from Illumina GenomeStudio or can be derived from Affymetrix CEL files, e.g. through the PennCNV libraries. The pipeline we use (and recommend) for Affymetrix SNP 6.0 arrays can be found within the R package on GitHub.
Please note that you need two adapted files for this pipeline, one containing the SNP locations for the AffySNP6 platform (12.68 MB, zip) and a genotype cluster file (33.25 MB, zip) that was compiled from a series of about 5,000 verified normal samples.
2) HTS data
For HTS data, ASCAT requires BAM files as well as reference files (listed on the GitHub page) so it can get allele counts and derive logR and BAF values. After logR/BAF files are generated (ascat.prepareHTS), one can use the other ASCAT functions to perform all of the standard steps (loading logR/BAF, correcting logR for covariates, segmenting tracks and getting CNA profiles).
For targeted sequencing data, we have implemented a bespoke function that identifies high-quality SNPs to investigate (ascat.prepareTargetedSeq). This step must be done on a batch of normal samples (no tumor samples) and prior to generating logR and BAF values. More information on how to get CNA profiles for HTS data can be found on our GitHub page.
3) Additional information
ASCAT can also be run on data from other species, for example, SNP arrays from canine breast cancers or exomes from zebrafish melanomas. As the method leverages SNP loci, it will however not work on haploid or homozygous (inbred) species (e.g. inbred mouse strains).
Samples profiled through SNP arrays or massively parallel sequencing are often affected by 'wave artifacts' that are in part correlated with the GC content of the surrounding region (e.g. this paper by Diskin et al.). We have implemented a GC wave correction in ASCAT, and recommend adding that step to the pipeline if the input data hasn't been through alternative methods for logR correction. Our original GC correction method (ASCAT 2.2) is based on the one initially implemented by Cheng et al., Genome Biology 12:R80, 2011. We have extended such a correction method to correct for both GC content and replication timing (as from version 3.0).
An important platform- and normalization-specific parameter is the normalization parameter (gamma) within the function ascat.runAscat. This parameter represents the drop in LogR for a change from two copies to one copy in 100% of cells. For massively parallel sequencing data, gamma should always be set to 1. For array data, due to array background signal and bespoke array normalization procedures, gamma is often significantly lower in practice. Its default setting of 0.55 works for many but not all SNP arrays (e.g. Illumina 109k arrays as processed through BeadStudio/GenomeStudio and Affymetrix SNP 6.0 arrays processed through the PennCNV libraries). For other SNP array platforms (and normalization procedures), we recommend checking the value of gamma through a comparison of a male and female germline sample (evaluating the difference in LogR values of the X chromosome probes between genders, relative to the rest of the genome), or through an X chromosome titration series.
ASCAT outputs
The output of ASCAT, and how to interpret it, is described in this book chapter.
Legacy versions and data
Historic versions of ASCAT are available as part of our GitHub version. We recommend always using the latest version, but we provide the historic versions for legacy reasons.
Major changes to ASCAT over the original version 1.0 are:
● Availability as an easy-to-use and coherent R software suite (2.0)
● Major improvements in computational speed (2.0)
● Platform-independence (2.0)
● Update of the core algorithm for better performance and results (2.0 and 2.2)
● Addition of germline genotype prediction and thereby extension to unmatched tumor samples (2.0)
● Adaptations to the ASPCF segmentation algorithm to increase sensitivity in samples with low noise and to increase robustness in more noisy samples (2.1)
● Addition of a gender parameter, allowing correct handling of copy number aberrations on the X chromosome in male samples (2.2)
● Addition of GC correction code (2.2)
● Adaptations to allow manual refitting of samples (2.3)
● Adaptations and additions to output data structures (2.3)
● Availability as R package (2.4)
● Addition of a multi-sample segmentation for samples that are expected to share breakpoints (2.5)
● Addition of a bespoke methodology for generating logR and BAF from HTS data (3.0)
● Addition of a pre-processing step for targeted sequencing data (3.1)
Breast carcinoma SNP array data from our original ASCAT publication is also available. The data consists of the LogR and BAF values for both the tumor and germline SNP array data. We also include tumor LogR data after adjustment for GC bias using the method described in Diskin et al., Nucleic Acids Research, 36:e126, 2008. Due to privacy regulations, the data are password protected. Please contact us to obtain access.
A script used to analyze these Illumina 109k breast carcinoma SNP array data using ASCAT 1.0 is available on GitHub.
Subclonal copy number analysis: the Battenberg algorithm
To assay subclonal copy number changes in massively parallel sequencing data, we created the Battenberg algorithm, based on the underlying ASCAT principles and equations and on haplotype phasing of 1000 genomes SNP loci. The Battenberg algorithm was originally described here and is now available on GitHub.
Frequently asked questions (FAQ)
Can I use ASCAT for (germline) CNV analysis?
ASCAT is a tool to detect somatic copy number alterations (CNAs) in cancer samples and cannot be applied to detect germline copy number variants (CNVs). The term CNV refers to a germline variant, polymorphic in a population. To avoid confusion, for somatic copy number changes in tumor samples, we recommend always using the term CNA.
Which version of ASCAT should I use?
We recommend always using the latest ASCAT version.
Can ASCAT be applied to cell lines as well?
ASCAT will work well on matched cell line data. However, it is not well suited to analyze unmatched cell line data, as the germline genotype prediction tool leverages the signal from admixed normal cells to infer germline genotypes. As most cell lines are in practice unmatched, ASCAT will most likely not be the ideal method for the analysis of cell line data.
When should I use ASCAT? When should I use Battenberg?
The Battenberg algorithm is specifically designed for detecting subclonal copy number changes on whole-genome sequencing data. The current version of Battenberg can also infer purity and ploidy from the data and would be our method of choice for the analysis of whole-genome sequencing data.
For analysis of other sequencing data (exome or targeted pulldown), haplotype phasing has more limited added value, and we recommend using ASCAT. ASCAT also supports the analysis of data from other species and the analysis of a wide range of SNP arrays.

About
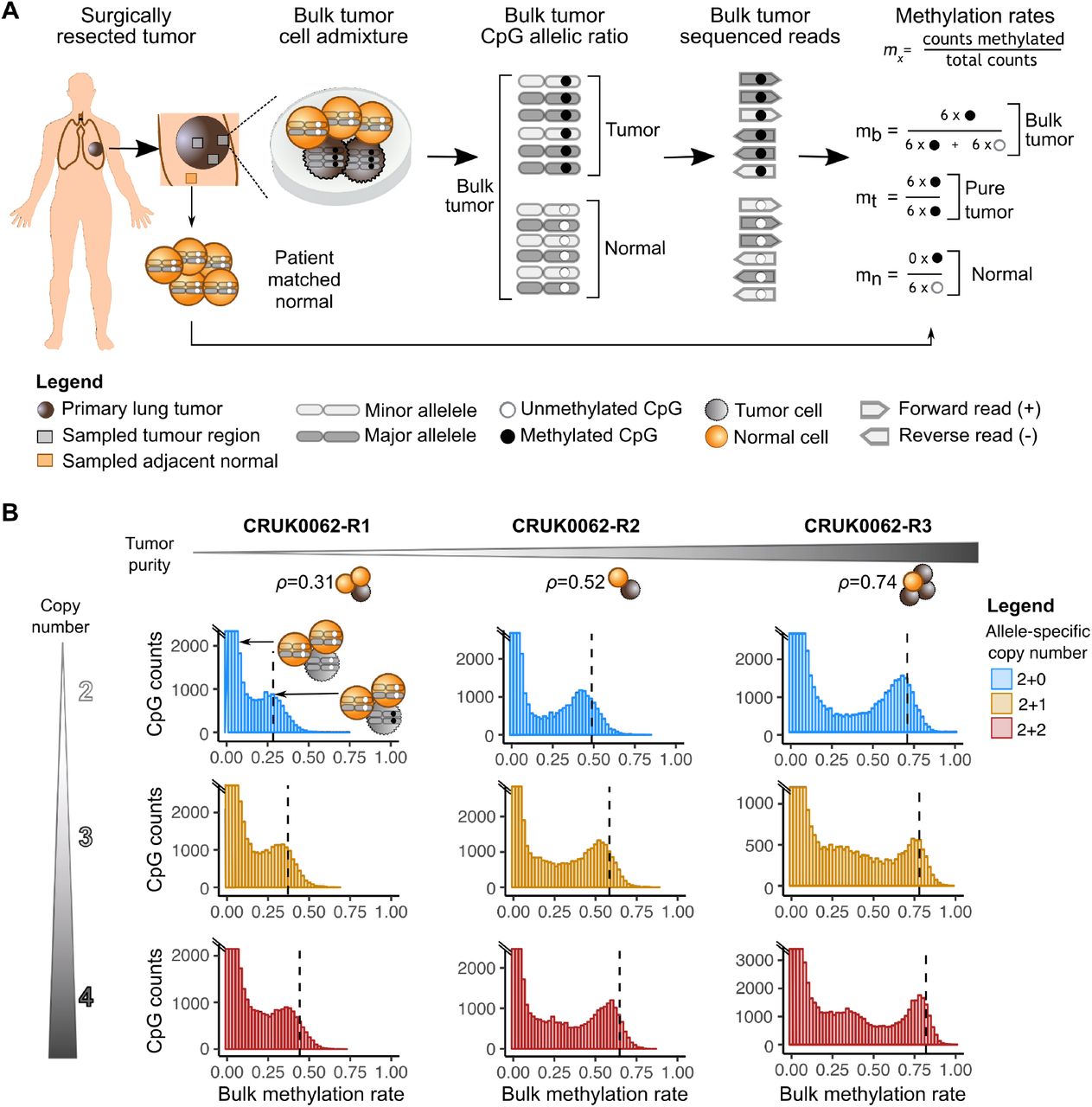
CAMDAC is a method for deconvolving bulk tumor bisulfite sequencing data. Bulk tumor DNA methylation is confounded by normal cell admixture and variable tumor DNA content driven by copy number alterations. CAMDAC infers tumor purity, ploidy, and allele-specific copy number states from bulk methylomes, combines these quantities with a reference for the bulk normal methylation fraction at each site, and derives the pure tumor methylation fraction per CpG site. We demonstrated that adjacent patient-matched adjacent normal tissue is a suitable proxy for the normal infiltrating cell population. CAMDAC-derived methylomes represent the average methylation per CpG allele in the tumor population and power the discovery of true tumor differential methylation events.
Resources
- CAMDAC github repository
- CAMDAC Documentation (RRBS / WGBS)
- CAMDAC preprint
Citation
If you use CAMDAC in your work, please cite:
Larose Cadieux, E., Mensah, N.E., Castignani, C., Tanić, M., Wilson, G.A., Dietzen, M., Dhami, P., Vaikkinen, H., Verfaillie, A., Cotobal Martin, C., Baker, T., Watkins, T.B.K., Selvaraju, V., Jamal-Hanjani, M., Kanu, N., McGranahan, N., Feber, A., TRACERx Consortium, Swanton, C., Beck, S., Demeulemeester, J., and Van Loo, P. 2022. Copy number-aware deconvolution of tumor-normal DNA methylation profiles. bioRxiv, doi: https://doi.org/10.1101/2020.11.03.366252.
CAMDAC
For a paired tumor-normal study design, CAMDAC accepts BAM files and proceeds through allele-counting, copy number calling, deconvolution and differential methylation testing. For example:
```
library(CAMDAC)
tumor = create_camdac_sample("P1_T1", bam_file = "tumor.bam")
normal = create_camdac_sample("P1_N1", bam_file = "normal.bam")
config = create_camdac_config(outdir="./results", bsseq="wgbs", bsseq_lib="pe", build="hg38")
pipeline_tumor_normal(tumor, normal, config)
```
CAMDAC results
CAMDAC returns several output files for each pipeline stage. Deconvolved methylation rates are found in a data frame stored in the R object `CAMDAC_results_per_CpG.RData`.
Important fields include:
• m_t: CAMDAC pure tumour methylation rate
• m_t_low: CAMDAC pure tumour methylation rate HDI99 lower boundary
• m_t_high: CAMDAC pure tumour methylation rate HDI99 upper boundary
• prob: Tumour-normal DMP probability
• CG_CN: CpG allele total copy number
• nA: Major allele copy number
• nB: Minor allele copy number
• segment: Copy number segment endpoints

Supported platforms
CAMDAC currently supports two platforms: reduced representation bisulfite sequencing (RRBS) and whole genome bisulfite sequencing (WGBS). CAMDAC has been verified with these platforms using the Bismark aligner.
Frequently asked questions (FAQ)
Where can I find information on CAMDAC outputs?
The CAMDAC output files are described at the end of the documentation for the respective platforms (RRBS / WGBS).
Can I use CAMDAC without matched normal tissue?
Yes, CAMDAC can be run without matched normal tissue by pooling sex- and tissue-matched methylation from patient samples, ideally with the same library preparation protocol and pre-processing pipeline.
Can I use CAMDAC for metastatic samples?
Yes, CAMDAC can be applied to metastatic tumors, but it is important to provide a normal methylation profile for both the infiltrating cells the cell of origin.
What is the best way to run CAMDAC for multi-region samples?
For multi-region samples, CAMDAC can use the same patient-matched normal tissue reference for each bulk tumor sample.
Image sources:
- Figure 1A is from CAMDAC paper, available at bioRxiv
- CAMDAC QC output example (from RRBS manual)
{kind=link}